Mitochondrial diseases are rare and complex genetic disorders that impair energy production, often leading to deficiencies in the oxidative chain. Respiratory chain complex III (CIII) deficiencies primarily manifest in the childhood and the affected patients develop diverse clinical symptoms.
In the present study, recently published in BBA – Molecular Basis of Disease, the research team of the German Mouse Clinic at the Helmholtz Center Munich together with collaborators from the University of Tampere and the University of Jena investigated a potential rescue strategy for complex III deficiencies. The alternative oxidase of C. intestinalis, AOX, was introduced into the Uqcrh KO mouse, a model of complex III disease, aiming to support the impaired respiratory chain.
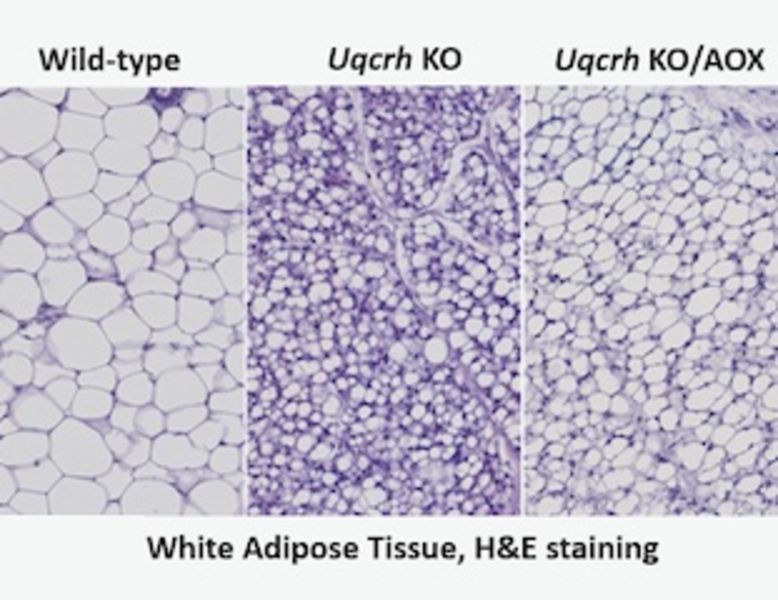
AOX acts as a bypass for the cytochrome segment of the mitochondrial respiratory chain when it is unavailable due to overload, damage, or mutation. Although it does not contribute directly to ATP production, it has been shown to modify and in some cases rescue phenotypes of respiratory-chain disease models. As previously published, the Uqcrh KO mouse model carries the same deletionin the complex III subunit of the mitochondrial respiratory chain as patients with a deletion in the UQCRH gene, and shares characteristics and biochemical features. Loss of complex III subunit Uqcrh in this model causes post-weaning growth failure with metabolic disturbance leading to lethality at ~9 weeks of age (Vidali et al, 2021).
After generating mice with homozygosity for the Uqcrh deletion combined with hemizygosity for ubiquitously expressed AOX, the Uqox mouse model was characterized and compared with Uqcrh homozygous mutant littermates lacking AOX, and with wild-type littermate controls. Metabolic assessment, functional tests, blood parameters and organs were investigated. Uqox mice expressing AOX as a transgene initially exhibited improved health and delayed phenotype onset by several weeks. However, Uqox mice developed a growth arrest and metabolic phenotype similar to the Uqcrh KO mice, but some physiological parameters showed improvements in the Uqox mice at the same age, suggesting a delay in symptomatic disease onset. The expression of AOX delayed lethality by ~3 weeks.
Although the gradual metabolic and physiological deterioration was not permanently reversed, the work indicates transient amelioration of many phenotypes by transgenic AOX observable by the delayed onset and progression of this respiratory chain disease. Although permanent rescue was not achieved in this study, the scientists got novel insights into the function of UQCRH and the nature of the physiological disturbances resulting from its deficiency.
Jacobs HT, Szibor M, Rathkolb B, da Silva-Buttkus P, Aguilar-Pimentel JA, Amarie OV, Becker L, Calzada-Wack J, Dragano N, Garrett L, Gerlini R, Hölter SM, Klein-Rodewald T, Kraiger M, Leuchtenberger S, Marschall S, Östereicher MA, Pfannes K, Sanz-Moreno A, Seisenberger C, Spielmann N, Stoeger C, Wurst W, Fuchs H, Hrabě de Angelis M, Gailus-Durner V. AOX delays the onset of the lethal phenotype in a mouse model of Uqcrh (complex III) disease. Biochim Biophys Acta Mol Basis Dis. 2023 May 23;1869(7):166760. doi: 10.1016/j.bbadis.2023.166760. Epub ahead of print. PMID: 37230398.